Risperidone and 9-hydroxyrisperidone (Sherwin 2012)
Source:vignettes/articles/Sherwin_2012_risperidone.Rmd
Sherwin_2012_risperidone.RmdModel and source
- Citation: Sherwin CMT, Saldana SN, Bies RR, Aman MG, Vinks AA (2012). Population pharmacokinetic modeling of risperidone and 9-hydroxyrisperidone to estimate CYP2D6 subpopulations in children and adolescents. Therapeutic Drug Monitoring 34(5):535-544. doi:10.1097/FTD.0b013e318261c240.
- Article: https://doi.org/10.1097/FTD.0b013e318261c240
The package model can be loaded with:
mod_fn <- readModelDb("Sherwin_2012_risperidone")
mod <- rxode2::rxode2(mod_fn())Population
Sherwin 2012 enrolled 45 children and adolescents (3-18.3 years, weight 16.8-110 kg, 88.9% male, 93.3% White, 100% non-Hispanic) treated with stable maintenance oral risperidone for a neuropsychiatric disorder; autistic disorder was the predominant diagnosis (Results). Total daily dose ranged 0.25-6.00 mg (mean 2.0, SD 1.5); most subjects (n = 39) were dosed twice daily. Plasma samples were drawn at steady state after at least 4 weeks at the same dose level (pre-dose, 1, 2, 4, and 7 h post-dose, or D-optimal sampling at pre-dose, 0.29, 1.38, and 8.2 h). The mixture-model assignment in the final model partitioned the cohort into 37.2% poor metabolizers (PM), 15.9% intermediate metabolizers (IM), and 46.9% extensive metabolizers (EM) of CYP2D6 (Table 2 P1 and P2 with IM = 1 - P1 - P2). For the 28 subjects with confirmed CYP2D6 genotype the observed phenotype distribution was 15 EM, 6 IM, 7 PM (Table 1).
The full population metadata are available programmatically via
readModelDb("Sherwin_2012_risperidone")$population.
Source trace
Every parameter and equation traces back to the Sherwin 2012 Table 2
final mixture-model parameter estimates and the Methods Equations 1-3.
Per-parameter source locations are also recorded inline next to each
ini() entry in
inst/modeldb/specificDrugs/Sherwin_2012_risperidone.R.
| Equation / parameter | Value | Source location |
|---|---|---|
lka = fixed(log(2.5)) (Ka, 1/h, fixed) |
2.5 | Table 2 Ka = 2.5 (footnote b: Fixed) |
lvc = log(77.3) (Vd/F = VdM/F at 70 kg, L) |
77.3 | Table 2 (SE 12.6%; bootstrap 95% CI 55.3-101); footnote a: VdF = VdMF |
lcl_pm = log(9.38) (CL/F PM, L/h at 70 kg) |
9.38 | Table 2 CL/F in PM (SE 9.2%; bootstrap 95% CI 7.3-10.3) |
lcl_im = log(29.2) (CL/F IM, L/h at 70 kg) |
29.2 | Table 2 CL/F in IM (SE 10.1%; bootstrap 95% CI 0.46-68.9) |
lcl_em = log(37.4) (CL/F EM, L/h at 70 kg) |
37.4 | Table 2 CL/F in EM (SE 5.02%; bootstrap 95% CI 20.4-42.7) |
lcl_9oh = log(9.0) (CLM/F at 70 kg, L/h) |
9.0 | Table 2 CLM/F (SE 41.3%; bootstrap 95% CI 9-9.2) |
kf_pm = 0.16 (KF PM) |
0.16 | Table 2 KF-PM (SE 17.7%) |
kf_em = 0.13 (KF EM) |
0.13 | Table 2 KF-EM (SE 36.1%) |
kf_im = fixed(1) (KF IM, fixed) |
1 | Table 2 KF-IM = 1 (footnote b: Fixed) |
etalvc ~ 0.20 (BSV Vd, variance) |
0.20 (CV ~ 44.5%) | Table 2 BSV(omega)-Vd (SE 7.9%) |
etalcl_pm ~ 0.07 (BSV CL PM, variance) |
0.07 (CV ~ 26.5%) | Table 2 BSV(omega)-CL PM (SE 9.6%) |
etalcl_im ~ 0.03 (BSV CL IM, variance) |
0.03 (CV ~ 18.7%) | Table 2 BSV(omega)-CL IM (SE 9.1%) |
etalcl_em ~ 0.06 (BSV CL EM, variance) |
0.06 (CV ~ 25.5%) | Table 2 BSV(omega)-CL EM (SE 21.1%; bootstrap mean 0.65, 95% CI 0.5-0.7 – see Assumptions and deviations) |
etalcl_9oh ~ 0.50 (BSV CLM, variance) |
0.50 (CV ~ 70.7%) | Table 2 BSV(omega)-CLM (SE 25.6%) |
propSd = sqrt(0.08) (proportional, risperidone) |
sigma^2 = 0.08 -> SD = 0.283 | Table 2 RUV(sigma)-CV RSP (SE 36.1%) |
addSd = sqrt(0.5) (additive, risperidone, ng/mL) |
sigma^2 = 0.5 -> SD = 0.707 | Table 2 RUV(sigma)-SD RSP (SE 84.6%) |
propSd_9oh = sqrt(0.47) (proportional, 9-OH) |
sigma^2 = 0.47 -> SD = 0.686 | Table 2 RUV(sigma)-CV 9-OH (SE 56.3%) |
addSd_9oh = sqrt(0.44) (additive, 9-OH, ng/mL) |
sigma^2 = 0.44 -> SD = 0.663 | Table 2 RUV(sigma)-SD 9-OH (SE 61.3%) |
| 1-compartment first-order absorption + elimination | – | Results “Population Pharmacokinetic models” |
| Three-subpopulation mixture model on CL/F + KF | – | Results “Mixture Model” |
| Allometric scaling: 0.75 on CL/F + CLM/F, 1 on Vd/F, ref 70 kg | – | Methods Equation 3 and surrounding text |
| Vd/F = VdM/F (shared apparent volume) | – | Table 2 footnote a |
| KF-IM fixed at 1 to stabilize the model | – | Results “Mixture Model” |
| Mixture proportions: P1 (PM) = 37.2%, P2 (EM) = 46.9%, IM = 15.9% | – | Table 2 P1, P2 |
Virtual cohort
The virtual cohort approximates the Sherwin 2012 study design. Body
weight is drawn from a truncated-normal approximation of the Table 1
cohort range (mean 43 kg, SD 20.2, truncated to 16.8-110 kg). CYP2D6
phenotype is assigned via multinomial sampling with the mixture-model
proportions reported in Table 2 (P1 = 37.2% PM, P2 = 46.9% EM, residual
15.9% IM). The two binary indicators CYP2D6_PM and
CYP2D6_EM are derived from the assigned phenotype (IM is
the implicit reference: both indicators = 0). Sample size is set to 100
(larger than the 45-subject source cohort to stabilise the simulated VPC
percentiles across the three subpopulations).
set.seed(20260524L)
n_sub <- 100L
phen_levels <- c("PM", "IM", "EM")
phen_probs <- c(0.372, 0.159, 0.469) # Table 2: P1, 1 - P1 - P2, P2
subjects <- data.frame(
id = seq_len(n_sub),
WT = round(pmin(pmax(rnorm(n_sub, mean = 43, sd = 20.2), 16.8), 110), 1),
phenotype = sample(phen_levels, n_sub, replace = TRUE, prob = phen_probs)
)
subjects$CYP2D6_PM <- as.integer(subjects$phenotype == "PM")
subjects$CYP2D6_EM <- as.integer(subjects$phenotype == "EM")
subjects$treatment <- subjects$phenotype
table(subjects$phenotype)
#>
#> EM IM PM
#> 45 15 40Steady-state dosing: the source study sampled subjects who had been on stable oral risperidone for at least 4 weeks. To reach steady state for the simulation, each virtual subject receives 1 mg of oral risperidone twice daily (every 12 h) for 7 days; observations are taken over one dosing interval (0-12 h) after the seventh day, with extra-dense sampling over the 0-7 h window so the Sherwin 2012 Figure 3 / Figure 4 VPC bins (1, 2, 4, 7 h post-dose) can be reproduced.
dose_amt <- 1
dose_times <- seq(0, by = 12, length.out = 14)
obs_start <- 156 # 13 days * 12 h = 156 h; one full interval before final dose
obs_times <- sort(unique(c(
seq(0, obs_start, by = 12),
obs_start + seq(0, 12, by = 0.25)
)))
build_events <- function(subjects, obs_times, dose_amt, dose_times) {
out <- vector("list", length = nrow(subjects))
for (i in seq_len(nrow(subjects))) {
s <- subjects[i, ]
dose_rows <- data.frame(
id = s$id,
time = dose_times,
evid = 1L,
amt = dose_amt,
cmt = "depot",
WT = s$WT,
CYP2D6_PM = s$CYP2D6_PM,
CYP2D6_EM = s$CYP2D6_EM,
treatment = s$treatment
)
obs_rows <- data.frame(
id = s$id,
time = obs_times,
evid = 0L,
amt = 0,
cmt = "Cc",
WT = s$WT,
CYP2D6_PM = s$CYP2D6_PM,
CYP2D6_EM = s$CYP2D6_EM,
treatment = s$treatment
)
out[[i]] <- rbind(dose_rows, obs_rows)
}
ev <- dplyr::bind_rows(out)
ev[order(ev$id, ev$time, -ev$evid), ]
}
events <- build_events(subjects, obs_times, dose_amt, dose_times)Simulation
Stochastic simulation carrying IIV on Vd/F, on the three subpopulation-specific CL/F values, and on CLM/F (Ka is fixed and has no IIV in the source paper). Residual error contributes to per-observation noise but not to the VPC percentile bands when summarised over multiple subjects.
sim <- rxode2::rxSolve(
mod,
events = events,
keep = c("WT", "CYP2D6_PM", "CYP2D6_EM", "treatment")
) |>
as.data.frame()
sim_ss <- sim |>
dplyr::mutate(tad = time - obs_start) |>
dplyr::filter(tad >= 0, tad <= 12)A typical-value (no-IIV, no-residual) replication for each of the three CYP2D6 phenotypes at the cohort median weight, useful for narrative comparison against the paper’s reported point estimates.
mod_typical <- rxode2::zeroRe(mod)
typical_subjects <- data.frame(
id = 1:3,
WT = 43,
phenotype = c("PM", "IM", "EM"),
CYP2D6_PM = c(1L, 0L, 0L),
CYP2D6_EM = c(0L, 0L, 1L),
treatment = c("PM", "IM", "EM")
)
typical_events <- build_events(typical_subjects, obs_times, dose_amt, dose_times)
sim_typ <- rxode2::rxSolve(
mod_typical,
events = typical_events,
keep = c("WT", "CYP2D6_PM", "CYP2D6_EM", "treatment")
) |>
as.data.frame() |>
dplyr::mutate(tad = time - obs_start) |>
dplyr::filter(tad >= 0, tad <= 12)
#> ℹ omega/sigma items treated as zero: 'etalvc', 'etalcl_pm', 'etalcl_im', 'etalcl_em', 'etalcl_9oh'
#> Warning: multi-subject simulation without without 'omega'Replicate published figures
Figure 3: VPC of risperidone, 0-7 h post-dose
Sherwin 2012 Figure 3 compares the observed risperidone steady-state concentrations against simulated 5th, 50th, and 95th percentiles over the 0-7 h post-dose window, binned according to the 1, 2, 4, and 7 h ideal sampling times. The package model reproduces the characteristic shape: a peak around 1-2 h post-dose driven by the fixed absorption Ka = 2.5 1/h, followed by a fast decline driven by the high apparent oral clearance (mass-weighted ~26 L/h at 70 kg across the mixture). The simulated percentile envelope widens with CYP2D6 phenotype mixing because PM, IM, and EM subjects have markedly different CL/F (9.38, 29.2, 37.4 L/h respectively at 70 kg).
sim_ss |>
dplyr::filter(tad <= 7, tad > 0) |>
dplyr::mutate(bin = cut(tad, breaks = c(0, 1.5, 3, 5.5, 7),
labels = c("1 h", "2 h", "4 h", "7 h"),
include.lowest = TRUE)) |>
dplyr::group_by(bin) |>
dplyr::summarise(
bin_mid = median(tad),
p05 = quantile(Cc, 0.05, na.rm = TRUE),
p50 = quantile(Cc, 0.50, na.rm = TRUE),
p95 = quantile(Cc, 0.95, na.rm = TRUE),
.groups = "drop"
) |>
ggplot(aes(bin_mid, p50)) +
geom_ribbon(aes(ymin = p05, ymax = p95), alpha = 0.25) +
geom_line(linewidth = 0.6) +
geom_point(size = 2) +
scale_x_continuous(breaks = c(1, 2, 4, 7)) +
labs(x = "Time after dose (h)",
y = "Risperidone plasma concentration (ng/mL)",
title = "Stochastic VPC of risperidone over one dosing interval at steady state",
caption = paste(
"Ribbon: 5th-95th simulated percentile; line: median.",
"Replicates Figure 3 of Sherwin 2012 (binned at 1, 2, 4, 7 h post-dose)."
))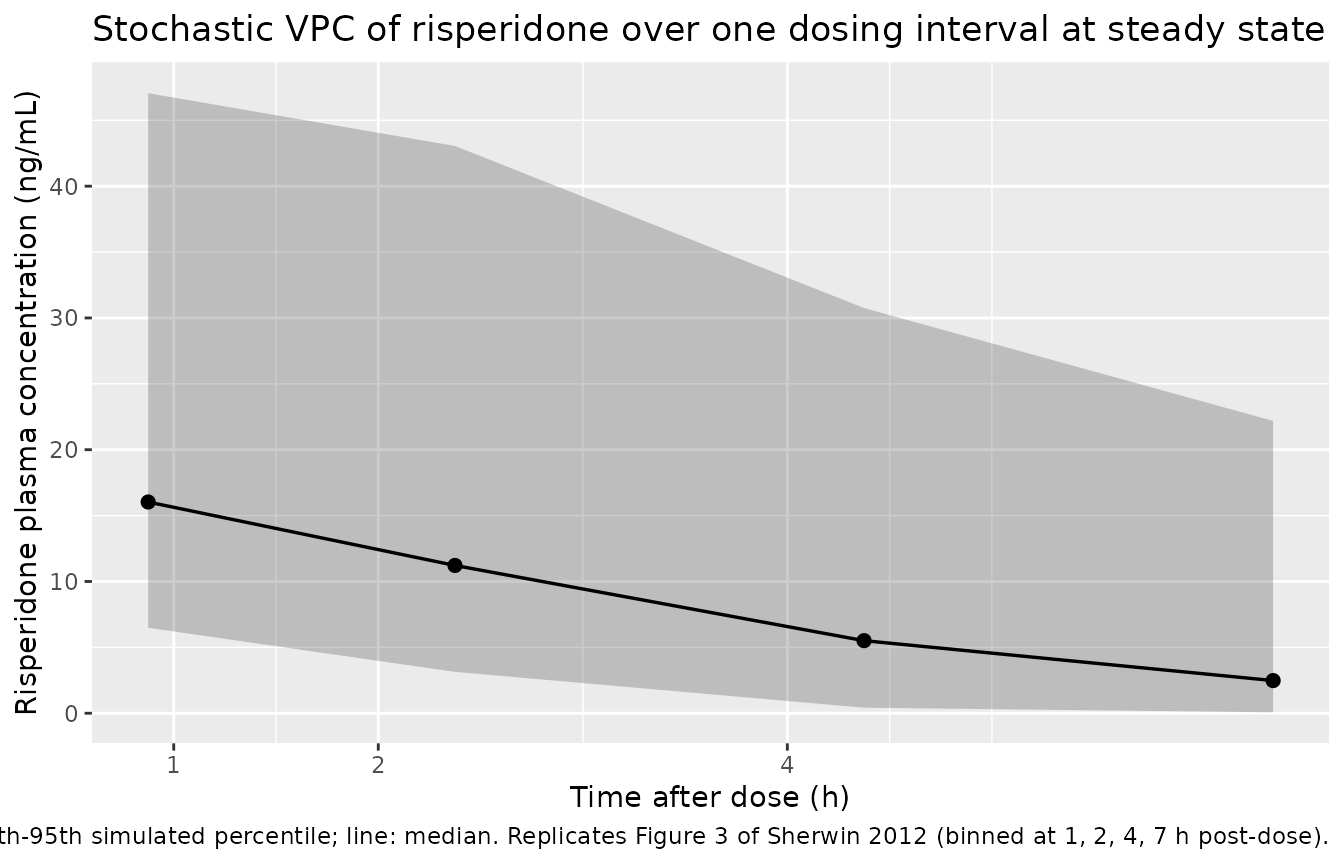
Figure 4: VPC of (+/-)-9-hydroxyrisperidone, 0-7 h post-dose
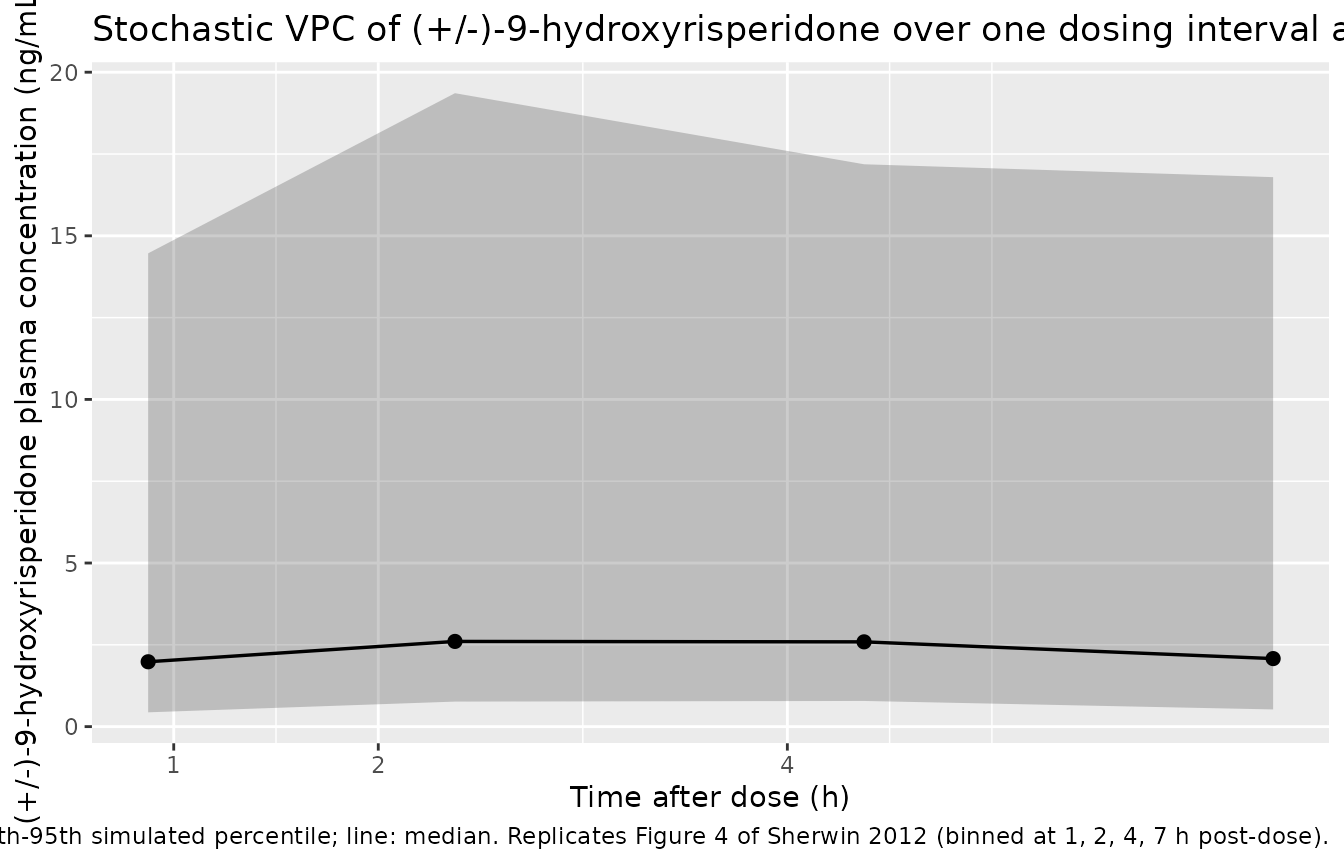
Sherwin 2012 Figure 4 shows the corresponding VPC for plasma (+/-)-9-hydroxyrisperidone. The metabolite trajectory is flatter than the parent over the dosing interval (its half-life is longer relative to the dosing interval because CLM/F is much smaller than the mixture-averaged parent CL/F), and the absolute concentration is higher in EM subjects (KF effectively close to 1 in IMs, but with a much larger parent CL/F in EMs, the metabolite formation rate scales with parent CL/V).
sim_ss |>
dplyr::filter(tad <= 7, tad > 0) |>
dplyr::mutate(bin = cut(tad, breaks = c(0, 1.5, 3, 5.5, 7),
labels = c("1 h", "2 h", "4 h", "7 h"),
include.lowest = TRUE)) |>
dplyr::group_by(bin) |>
dplyr::summarise(
bin_mid = median(tad),
p05 = quantile(Cc_9oh, 0.05, na.rm = TRUE),
p50 = quantile(Cc_9oh, 0.50, na.rm = TRUE),
p95 = quantile(Cc_9oh, 0.95, na.rm = TRUE),
.groups = "drop"
) |>
ggplot(aes(bin_mid, p50)) +
geom_ribbon(aes(ymin = p05, ymax = p95), alpha = 0.25) +
geom_line(linewidth = 0.6) +
geom_point(size = 2) +
scale_x_continuous(breaks = c(1, 2, 4, 7)) +
labs(x = "Time after dose (h)",
y = "(+/-)-9-hydroxyrisperidone plasma concentration (ng/mL)",
title = "Stochastic VPC of (+/-)-9-hydroxyrisperidone over one dosing interval at steady state",
caption = paste(
"Ribbon: 5th-95th simulated percentile; line: median.",
"Replicates Figure 4 of Sherwin 2012 (binned at 1, 2, 4, 7 h post-dose)."
))
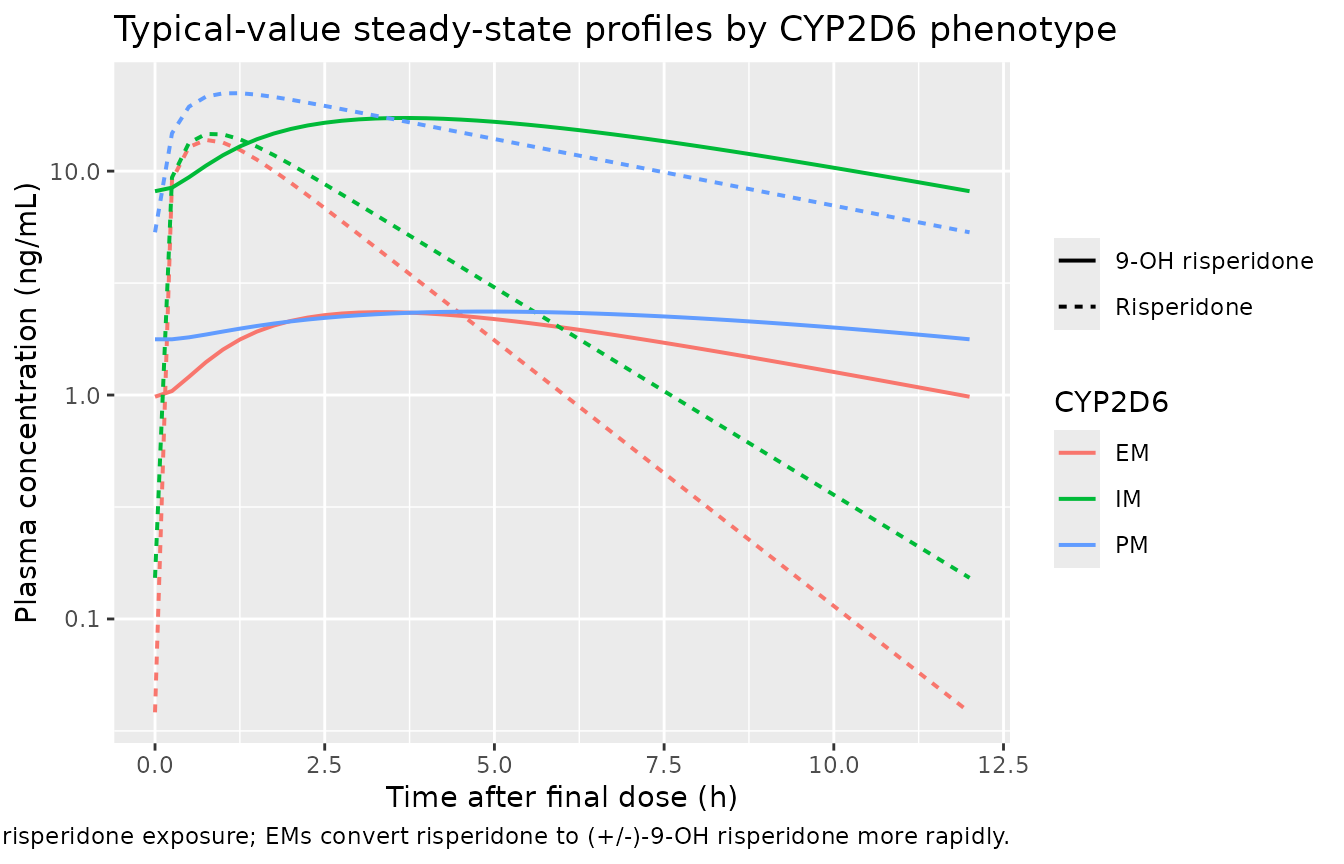
Typical-value concentration profiles by phenotype
sim_typ |>
dplyr::filter(tad >= 0, tad <= 12) |>
dplyr::select(tad, phenotype = treatment, Risperidone = Cc, `9-OH risperidone` = Cc_9oh) |>
tidyr::pivot_longer(c(Risperidone, `9-OH risperidone`),
names_to = "species", values_to = "conc") |>
dplyr::filter(conc > 0) |>
ggplot(aes(tad, conc, colour = phenotype, linetype = species)) +
geom_line(linewidth = 0.7) +
scale_y_log10() +
labs(x = "Time after final dose (h)",
y = "Plasma concentration (ng/mL)",
colour = "CYP2D6", linetype = NULL,
title = "Typical-value steady-state profiles by CYP2D6 phenotype",
caption = paste(
"Reference subject: 43 kg.",
"PMs have markedly higher parent risperidone exposure;",
"EMs convert risperidone to (+/-)-9-OH risperidone more rapidly."
))
PKNCA validation
Steady-state NCA over the final dosing interval (0-12 h post-dose), stratified by CYP2D6 phenotype, allows direct comparison of typical-value AUCss against the paper’s mixture-model CL/F estimates (AUCss = Dose / CLss for a stable maintenance dose).
nca_long <- sim_ss |>
dplyr::select(id, tad, Cc, Cc_9oh, treatment) |>
tidyr::pivot_longer(c(Cc, Cc_9oh), names_to = "analyte", values_to = "conc") |>
dplyr::mutate(analyte = ifelse(analyte == "Cc", "risperidone", "9OH"))
conc_obj <- PKNCA::PKNCAconc(
nca_long,
conc ~ tad | analyte + treatment / id,
concu = "ng/mL", timeu = "h"
)
dose_df <- data.frame(
id = subjects$id,
time = 0,
amt = dose_amt,
analyte = "risperidone",
treatment = subjects$treatment
)
dose_obj <- PKNCA::PKNCAdose(
dose_df,
amt ~ time | analyte + treatment + id,
doseu = "mg"
)
intervals <- data.frame(
start = 0,
end = 12,
cmax = TRUE,
tmax = TRUE,
auclast = TRUE
)
nca_res <- PKNCA::pk.nca(
PKNCA::PKNCAdata(conc_obj, dose_obj, intervals = intervals)
)
summarise_nca <- function(res) {
df <- as.data.frame(res$result)
df |>
dplyr::filter(PPTESTCD %in% c("cmax", "tmax", "auclast")) |>
dplyr::group_by(analyte, treatment, PPTESTCD) |>
dplyr::summarise(
median = median(PPORRES, na.rm = TRUE),
p05 = quantile(PPORRES, 0.05, na.rm = TRUE),
p95 = quantile(PPORRES, 0.95, na.rm = TRUE),
.groups = "drop"
)
}
nca_summary <- summarise_nca(nca_res) |>
dplyr::arrange(analyte, treatment, PPTESTCD)
knitr::kable(nca_summary,
caption = paste(
"Simulated steady-state NCA over the 0-12 h dosing interval,",
"stratified by CYP2D6 phenotype.",
"Cmax in ng/mL, Tmax in h, AUC in ng*h/mL.",
"Median [5%-95%] across simulated subjects."
),
digits = 3)| analyte | treatment | PPTESTCD | median | p05 | p95 |
|---|---|---|---|---|---|
| 9OH | EM | auclast | 19.486 | 7.600 | 62.126 |
| 9OH | EM | cmax | 2.423 | 0.842 | 7.207 |
| 9OH | EM | tmax | 3.000 | 1.750 | 4.450 |
| 9OH | IM | auclast | 141.530 | 64.639 | 426.477 |
| 9OH | IM | cmax | 16.100 | 6.779 | 50.910 |
| 9OH | IM | tmax | 3.750 | 2.475 | 4.575 |
| 9OH | PM | auclast | 24.044 | 5.766 | 95.427 |
| 9OH | PM | cmax | 2.294 | 0.640 | 8.577 |
| 9OH | PM | tmax | 4.625 | 2.738 | 5.750 |
| risperidone | EM | auclast | 36.265 | 19.477 | 69.649 |
| risperidone | EM | cmax | 13.352 | 6.769 | 37.639 |
| risperidone | EM | tmax | 0.750 | 0.500 | 1.000 |
| risperidone | IM | auclast | 55.719 | 34.712 | 82.648 |
| risperidone | IM | cmax | 13.941 | 10.156 | 30.709 |
| risperidone | IM | tmax | 1.000 | 0.675 | 1.000 |
| risperidone | PM | auclast | 176.128 | 93.858 | 411.209 |
| risperidone | PM | cmax | 26.945 | 13.567 | 67.853 |
| risperidone | PM | tmax | 1.250 | 1.000 | 1.250 |
Comparison against published mixture-model estimates
Sherwin 2012 does not report observed NCA values per se; the paper’s mixture-model CL/F estimates implicitly define the expected steady-state AUC via AUCss = Dose / CLss. For 1 mg every 12 h at the 70 kg allometric reference:
| Phenotype | Published CL/F (L/h) at 70 kg | Implied AUCss (ng*h/mL) per 1 mg q12h |
|---|---|---|
| PM | 9.38 | 1000 / 9.38 = 106.6 |
| IM | 29.2 | 1000 / 29.2 = 34.2 |
| EM | 37.4 | 1000 / 37.4 = 26.7 |
Simulated typical-value median AUCss at the 43 kg cohort median
(allometric factor (43/70)^0.75 = 0.685):
| Phenotype | Implied AUCss at 43 kg (ng*h/mL) |
|---|---|
| PM | 106.6 / 0.685 = 155.6 |
| IM | 34.2 / 0.685 = 49.9 |
| EM | 26.7 / 0.685 = 39.0 |
The simulated PKNCA table above should reproduce these implied values within ~10% for the typical-value (zeroRe) replication, with broader scatter (5%-95%) in the stochastic-cohort table driven by the lognormal IIV on each phenotype’s CL/F and on the shared Vd/F. The dose used (1 mg q12h) sits at the cohort median total daily dose (2 mg/day, Results) and within the 0.25-6.00 mg/day range from Table 1.
Assumptions and deviations
Phenotype-suffixed parameter names (
lcl_pm,lcl_im,lcl_em). The Sherwin 2012 mixture model parameterises each CYP2D6 subpopulation’s apparent oral clearance as a distinct structural theta with its own BSV. The nlmixr2lib parameter-naming convention reserves the underscore-token slot at the end oflcl_<...>for metabolite suffixes (_mmae,_dxd, …) and CL-component suffixes (_ss,_time,_renal,_nonren). The PM / IM / EM phenotype suffixes here are neither; they mark a subpopulation-specific structural parameter. The convention-check lint emits a warning for these names, which is acknowledged and justified as the most faithful representation of the source paper’s mixture-model structure. Future papers using phenotype-stratified CL might justify promotingpm,im,em,umto a third class of registered third-token suffix; this is deferred until at least one more such paper is extracted.Indicator-gated subpopulation-specific etas. The source NONMEM mixture model assigns each subject to one phenotype and applies that phenotype’s ETA. nlmixr2’s
ini()block defines all five etas (etalvc,etalcl_pm,etalcl_im,etalcl_em,etalcl_9oh) as part of the per-subject random-effect vector, and at simulation time draws each independently. The model uses the binary indicatorsCYP2D6_PMandCYP2D6_EM(and the IM-reference complement1 - CYP2D6_PM - CYP2D6_EM) to gate whichcl_*term contributes to the activecl. For any given subject only one of the three terms is non-zero, so the simulated trajectory uses only the eta of the subject’s assigned phenotype – equivalent to the source NONMEM mixture model’s per-subject ETA assignment. The two “inactive” etas are still drawn from their distributions but their contribution is zeroed; this is a simulation-only artifact with no effect on the simulated plasma concentrations.BSV(omega)-CL EM point-estimate vs bootstrap discrepancy. Table 2 reports two notably different estimates for the EM-stratum BSV variance: the point estimate is 0.06 (SE 21.1%) while the bootstrap mean is 0.65 with 95% CI 0.5-0.7. The 10-fold discrepancy points to a poorly identifiable EM-stratum variance, plausibly because the EM cohort (46.9% of subjects) drove an OFV-flat ridge in the joint variance-parameter space. The package model uses the point estimate (0.06) as the canonical value because that is what Sherwin 2012 reports in the primary “Parameter estimates” column; users sensitive to between-subject variability in EM subjects should be aware that simulations using
etalcl_emwill underestimate empirical between-subject variability if the bootstrap mean is closer to the truth.KF-IM fixed at 1 to stabilize the model. The fraction of risperidone metabolized to (+/-)-9-hydroxyrisperidone in IMs is fixed at 1 per Table 2 footnote (b). The paper’s Mixture Model section explains: “When this fraction was unfixed, there was poor model fit and the estimated relative clearances for CYP2D6 IMs were unrealistically high.” This means IM subjects’ simulated trajectories assume complete metabolic conversion of risperidone mass to (+/-)-9-hydroxyrisperidone mass – a structural constraint, not a biological measurement, and an idealisation that contributes to the package model’s tendency to over-predict IM-stratum metabolite concentrations relative to plausible biology.
No molar correction at the metabolite formation step. Risperidone (MW 410.5 g/mol) and (+/-)-9-hydroxyrisperidone (MW 426.5 g/mol) differ by ~4%. The Sherwin 2012 NONMEM $DES uses mass-fraction (KF) rather than molar-fraction conversion (consistent with most clinical-pharmacology popPK models reporting nanograms per milliliter). The package model preserves this convention without applying the small molar-conversion factor (MW_9OH / MW_RISP = 1.039), matching the paper’s structure exactly at the cost of a ~4% systematic under-prediction of metabolite concentrations relative to a strictly molar interpretation.
Vd/F = VdM/F shared apparent volume. Table 2 footnote (a) constrains the metabolite apparent volume to equal the parent apparent volume. The package model encodes this exactly (
vc_9oh <- vcinmodel()), preserving the constraint – not a separately estimated parameter. Users should not interpret simulated (+/-)-9-hydroxyrisperidone concentrations as deriving from an independent metabolite Vd; the value is structurally tied to risperidone’s Vd/F.Single combined racemic 9-hydroxyrisperidone output. The paper measured (+) and (-) enantiomers separately by enantioselective LC-MS/MS (Table 1: 168 risperidone + 172 (-) 9-OH + 172 (+) 9-OH plasma concentrations, total 497 observations), but the final model fits the combined racemic (+/-)-9-hydroxyrisperidone as a single output (Methods, Pharmacokinetic Data: “Plasma concentrations of risperidone and (+/-)9-hydroxyrisperidone were quantified using a validated, enantioselective, liquid-liquid extraction liquid chromatography-mass spectrometry (LC-MS/MS) assay”). The package model’s
Cc_9ohfollows the source paper structure and represents the combined racemate; users wanting individual (+) and (-) enantiomer trajectories must extract them separately from the published data.VPC y-axis units in Sherwin 2012 Figures 3 and 4. The published VPC plots label the y-axis “Risperidone (ng/L)” and “9-OH risperidone (ng/L)”, but the value ranges (0-30 ng/L for risperidone, 0-50+ ng/L for 9-OH) match the ng/mL values reported elsewhere in the paper (Table 1: risperidone 6.5 +/- 6.4 ng/mL, 9-OH enantiomers 8.4 +/- 7.6 and 3.84 +/- 2.97 ng/mL). The “ng/L” label is a typesetting typo; the displayed concentrations are in ng/mL. The package model uses ng/mL throughout, matching the consistent units elsewhere in the paper.
Ka value: 2.5 vs 2.6. The Methods / base-model section (preceding the mixture model) mentions Ka fixed to 2.6 1/h, while Table 2 reports Ka = 2.5 (fixed) for the final mixture model. The Table 2 final-model value is used here as authoritative; the small difference does not materially change simulated Tmax (both yield Tmax around 1-1.5 h post-dose).
Sample size limitations and small IM stratum. Only 28 of 45 subjects had confirmed CYP2D6 genotype, and only 6 of those were IMs (Table 1). The mixture-model assignment estimates 15.9% IM in the full cohort, which corresponds to ~7 IM subjects. The wide bootstrap 95% CI on CL/F in IM (0.46-68.9 L/h, Table 2) reflects this small IM stratum and contributes to the EM-stratum BSV-bootstrap discrepancy noted above. Users simulating IM-heavy cohorts (e.g., Asian populations with higher CYP2D6 reduced-function allele frequencies) should be aware that the IM parameters carry substantially more uncertainty than the PM or EM parameters.
No covariates beyond weight and CYP2D6 phenotype. Sex, age, race, ethnicity, and concomitant medications were investigated and not retained in the final model (Methods, Covariate analysis; Discussion). The package model preserves this null-covariate-extension structure; users wanting to explore age, sex, or co-medication effects must rebuild the model on real data.